I. The Primary Cilium and Common Disease
Obesity, kidney failure, diabetes, hepatic fibrosis – each of these diseases remains a major public health burden despite billions of dollars spent on their treatment and research every year. For patients with rare syndromic genetic disorders affecting the primary cilium the dysfunction of this single organelle can produce all these diseases in a single person. By studying the primary cilium, can we discover new molecular mechanisms of disease pathogenesis and identify novel therapeutic targets for common diseases of huge public health burden?
Found on nearly every human cell type, the primary cilium is a small, non-motile projection of the cell’s apical surface that acts as an antenna for the reception and integration of signals from the extracellular environment. A number of rare syndromic genetic disorders, termed ciliopathies, are known to result from perturbation of any of the ~300 genes critical to ciliary function. The pathologies characteristic of the ciliopathies include extreme risk of common diseases like kidney disease, diabetes, obesity, and hepatic fibrosis, which are caused by deficits in cell signaling pathways dependent on the cilium for normal function. Evidence now suggests that many signaling pathways with critical roles in common complex diseases pathogenesis (PDGF, TGFβ , SHH, Insulin) are dependent on the ciliary localization of their cognate receptors for appropriate downstream signaling.
Previous work in our lab has found that common genetic variants affecting ciliary gene expression are associated with markers of diabetes, liver function, and kidney health in the general population, with different sets of ciliary genes conveying increased risk for different diseases. This suggests that there exist distinct molecular mechanisms, each involved in different aspects of ciliary signaling/function, that play a role in the risk for common diseases like obesity and diabetes. However, despite these findings and despite the many links between ciliary signaling and pathways important to common disease pathogenesis, the cilium’s contribution to the development of common disease remains largely unexplored.

1. Identifying variants in ciliary genes that increase risk for common complex disease
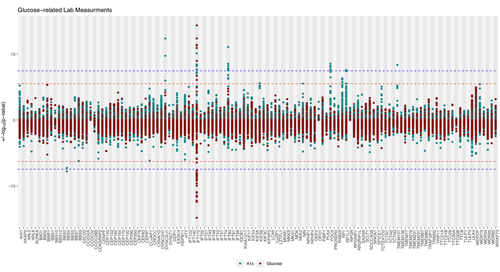
2. Differentiating the ciliary genes required for different disease-associated signaling pathways
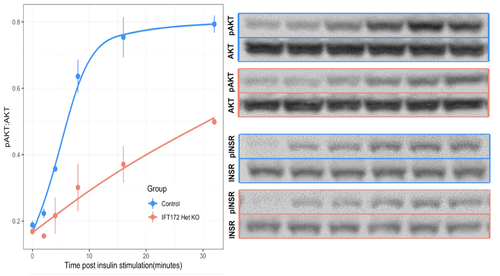
3. Comparing the effects of different chronic disease states on cilium structure and function

II. Genetic Variation and Disease on the Population Level
With the advent of larger and larger population-level biobanks linked to genetic data and the electronic health record, and with the development of more robust artifical intelegence approaches and more powerful statistical models, we have the opportunity to ask questions about the genetic architecture of human disease in ways never before possible. Our lab is actively exploring ways of integrating rare and common genetic variants to make fundamtental discoveries that begin to explain how our genes affect our health.
Our lab has shown that "rare" genetic disorders are actually much more common than previously thought, and often obscured by complex genetic paradigms such as somatic mosaicism, reduced penentrance, and unexpected pleiotropy. We have undertaken collaborative projects to begin to understand how burdens of rare deletarious variants in individual genes can influence a person's risk for disease, ranging from cardiomyopathy to COVID-19. We will continue to apply these approaches to further our knowledge of the rare genetic variant contribution to human disease phenotypes across the genome and phenome. There are projects available in this area for students, clinicians, and postdocs, with the diseases and genes to be investigated tailored to each person's interests.
But the big challenge that remains to be answered is this: can we integrate data on all of an individual's genetic variants -- both common and rare -- with our knowledge of gene pathways and interaction networks to more robustly predict each person's risk for disease? Can we use such a model to more precisely target our medications and interventions and make fundamental discoveries about the pathways involved in disease pathogenesis? We are working on AI-driven methods to do exactly this. We have many projects ongoing in this area, well-suited to scientists with computational and informatic interests and experience.

III. Genetics and Medicine:
Disparities, Implementation, and the Unknown
Disparities, Implementation, and the Unknown
It seems inevitable that over the next decade genomic data will become an integral component of each patient's medical record. Healthcare systems are investing heavily in Genetic and Precision Medicine initiatives that stand to fundamentally change the practice of medicine. But while there is immense promise in the implementation of these approaches, there are many important questions and hurdles that must be overcome to ensure the fair, effective, and economic implementation of genomic data in healthcare. Which patients will undergo genetic testing? Who will analyze and review the results? How can we harness this data to ensure that the right patient gets the right treatment at the right time?
Our lab, and others, has been working to understand how genetic data is currenlty being incorprated into clinical medicine so that we can prepare for and anticipate both the remarkable advances and major pitfalls that will come with these new technologies. Our data show very clearly that the current system amplifies and propagates race- and socioeconomic-status-based disparities, and our findings highlight major failings in our ability to identify patients who would benfit from genetic testing. Using the data derived from the electronic health record and from the deidentified genetic information available through the Penn Medicine BioBank, we can begin to identify and address these issues. We have many projects ongoing in this area, working to identify and address the hurdles that must be overcome to ensure the fair roll out of Precision and Genetic Medicine initiatives, and working to design and implement new approaches that will bring precision medicine to fruition in a just, conscientious, and economic way.

Obesity, kidney failure, diabetes, hepatic fibrosis – each of these diseases remains a major public health burden despite billions of dollars spent on their treatment and research every year. For patients with rare syndromic genetic disorders affecting the primary cilium the dysfunction of this single organelle can produce all these diseases in a single person. By studying the primary cilium, can we discover new molecular mechanisms of disease pathogenesis and identify novel therapeutic targets for common diseases of huge public health burden?
Found on nearly every human cell type, the primary cilium is a small, non-motile projection of the cell’s apical surface that acts as an antenna for the reception and integration of signals from the extracellular environment. A number of rare syndromic genetic disorders, termed ciliopathies, are known to result from perturbation of any of the ~300 genes critical to ciliary function. The pathologies characteristic of the ciliopathies include extreme risk of common diseases like kidney disease, diabetes, obesity, and hepatic fibrosis, which are caused by deficits in cell signaling pathways dependent on the cilium for normal function. Evidence now suggests that many signaling pathways with critical roles common complex diseases pathogenesis (PDGF, TGFβ , SHH, Insulin) are dependent on the ciliary localization of their cognate receptors for appropriate downstream signaling.
Previous work in our lab has found that common genetic variants affecting ciliary gene expression are associated with markers of diabetes, liver function, and kidney health in the general population, with different sets of ciliary genes conveying increased risk for different diseases. This suggests that there exist distinct molecular mechanisms, each involved in different aspects of ciliary signaling/function, that play a role in the risk for common diseases like obesity and diabetes. However, despite these findigfindings and despite the many links between ciliary signaling and pathways important to common disease pathogenesis, the cilium’s contribution to the devellopment of common disease remains largely unexplored.

1. Identifying variants in ciliary genes that increase risk for common complex disease
2. Differentiating the ciliary genes required for different disease-associated signaling pathway
3. Comparing the effects of different chronic disease states on cilium structure and function
While it is clear that dysfunction of the priamry cilium can have a profound impact on human health, little is known about the reciprocal effects of different common disease states on cilium structure and function. Recent studies and preliminary results from our lab have identified significant differences in ciliary gene expression between diabetic and non-diabetic mice and humans, suggesting that secondary defects in cilium function can result from common disease states. However, how these secondary changes interact with an individual's underlying genetics to exacerbate, or perhaps ameliorate, the symptoms of disease is unknown. Projects are available in this space, using RNAseq and microscopy to discover and define secondary changes in cilium function in animal models of common disease and human tissues.
With the advent of larger and larger population-level biobanks linked to genetic data and the electronic health record, and with the development of more robust artifical intelegence approaches and more powerful statistical models, we have the opportunity to ask questions about the genetic architecture of human disease in ways never before possible. Our lab is actively exploring ways of integrating rare and common genetic variants to make fundamtental discoveries that begin to explain how our genes affect our health.
Our lab has shown that "rare" genetic disorders are actually much more common than previously thought, and often obscured by complex genetic paradigms such as somatic mosaicism, reduced penentrance, and unexpected pleiotropy. We have undertaken collaborative projects to begin to understand how burdens of rare deletarious variants in individual genes can influence a person's risk for disease, ranging from cardiomyopathy to COVID-19. We will continue to apply these approaches to further our knowledge of the rare genetic variant contribution to human disease phenotypes across the genome and phenome. There are projects available in this area for students, clinicians, and postdocs, with the diseases and genes to be investigated tailored to each person's interests.
But the big challenge that remains to be answered is this: can we integrate data on all of an individual's genetic variants -- both common and rare -- with our knowledge of gene pathways and interaction networks to more robustly predict each person's risk for disease? Can we use such a model to more precisely target our medications and interventions and make fundamental discoveries about the pathways involved in disease pathogenesis? We are working on AI-driven methods to do exactly this. We have many projects ongoing in this area, well-suited to scientists with computational and informatic interests and experience.
II. Genetic Variation and Disease on the Population Level
III. Genetics and Medicine:
Disparities, Implementation, and the Unknown
It seems inevitable that over the next decade genomic data will become an integral component of each patient's medical record. Healthcare systems are investing heavily in Genetic and Precision Medicine initiatives that stand to fundamentally change the practice of medicine. But while there is immense promise in the implementation of these approaches, there are many important questions and hurdles that must be overcome to ensure the fair, effective, and economic implementation of genomic data in healthcare. Which patients will undergo genetic testing? Who will analyze and review the results? How can we harness this data to ensure that the right patient gets the right treatment at the right time?
Our lab, and others, has been working to understand how genetic data is currenlty being incorprated into clinical medicine so that we can prepare for and anticipate both the remarkable advances and major pitfalls that will come with these new technologies. Our data show very clearly that the current system amplifies and propagates race- and socioeconomic-status-based disparities, and our findings highlight major failings in our ability to identify patients who would benfit from genetic testing. Using the data derived from the electronic health record and from the deidentified genetic information available through the Penn Medicine BioBank, we can begin to identify and address these issues. We have many projects ongoing in this area, working to identify and address the hurdles that must be overcome to ensure the fair roll out of Precision and Genetic Medicine initiatives, and working to design and implement new approaches that will bring precision medicine to fruition in a just, conscientious, and economic way.